特发性肺纤维化(idiopathicpulmonaryfibrosis,简称IPF),亦称隐源性纤维化肺泡炎或特发性间质性肺炎等。本病病因未明,弥散性肺间质纤维化局限于肺部。Hamman和Rich分别于1935年和1944年首先报道数例,均属急性型,进展急剧,在6周至半年内死亡,故又名Hamman-Rich综合症。本病多在40-50岁发病,男性稍多于女性,是遍及全世界的疾病,近10年来发病率及病死率皆升高,绝大多数为慢性型,发病年龄越高,病程越长。急性型罕见。近年多数学者认为系自身免疫性疾病,可能与遗传因素有关。
[病理]
肺病理变化以肺泡壁细胞浸润、增厚、间质纤维化为特点。肉眼观察肺脏质地坚实,有橡胶触感,有大小不等的囊肿凸出于肺表面,并有交错分布的灰白色纤维素条和瘢痕。镜检示肺组织结构有变形,肺泡表面细胞肿胀,呈正方或短柱形,肺泡壁增厚,内有灶性出血及成纤维细胞增殖。肺泡腔内有来自肺泡的单核细胞、组织细胞、嗜酸粒细胞及渗出液。间质呈水肿、有单核细胞、淋巴及少量浆细胞浸润,随后细长成纤维细胞和大量胶原纤维出现,部分间质腔几乎完全纤维化。晚期肺泡数量明显减少、变形、闭锁或残留裂隙状不规则形态。细支气管代偿、扩张成蜂窝肺。受累肺脏由於大量纤维结缔组织增殖而收缩,毛细血管数量减少甚至闭锁。病情持续发展,肺组织病理改变亦不停地由早期向晚期发展,故在同一视野中可看到各阶段的病理变化,即肺泡炎、肺泡结构紊乱和蜂窝肺。
[临床表现]
以隐袭性进行性呼吸困难为其突出的症状,体力衰弱,可有盗汗、食欲减退、体重减轻、消瘦、无力等。轻度干咳、偶有痰血。体检:呼吸浅速,两肺底吸气期啰音似Velcro尼龙带拉开音。50%患者有杵状指(趾)和紫绀。晚期病例右心受累,显示肺心病症状和体征,最后多死于呼吸、循环衰竭。
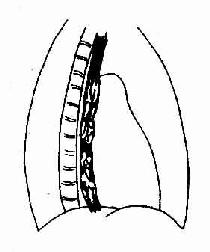
早期虽有呼吸困难症状,但X线胸片可能基本正常;中后期可出现两肺中下野弥散性网状或结节状阴影,偶见胸膜腔积液、增厚或钙化。
肺功能表现为进行性限制性通气功能障碍和弥散量减少。
实验室检查示血乳酸脱氢酶增高;类风湿因子、抗核抗体和丙种球蛋白可增高。
[治疗]
在急性病例用糖皮质激素,通常泼尼松0.5-1mg/kg.d,分3次服用,1-2个月,待主观和客观指标(X线和肺功能)达最佳水平,不断继续好转时,逐渐减量至25mg/d,晨15mg,下午10mg,维持2个月,再减至20mg/d,分两次服用2-3个月。20mg/d后每次减量2-5mg.最后维持量为5-7.5mg/d,空腹一次服用,疗程不少于1年。但在治疗期间,要注意并发肺结核,应及时诊治。
老年病人,发绀、杵状指、低氧血症,胸片示广泛肺纤维化,蜂窝肺者,只对症治疗,吸氧,不宜用糖皮质激素。皮质激素无效或不能接受者,可应用免疫抑制剂如小剂量硫唑嘌呤。
继发感染常是病情恶化、导致死亡的因素,故预防和及时控制呼吸道感染是治疗中不可忽视的环节。