概述
粘多糖病(mucopolysacharidosis,MPS)是因酸性粘多糖降解酶缺乏,使之不能完全降解,其产物在体内堆积所致。由于各种成分在体内分布的不同,以及不同酶的缺乏,粘多糖病在临床上表现亦各异,多以骨骼病变为主,还可累及中枢神经系统、心血管系统以及肝、脾、关节、肌腱、皮肤等。按其代谢产物和临床表现共分为8型,有的还有数种亚型。
病因和发病机制
粘多糖(macopolg sacrotein MPS)是骨基质和结缔组织的主要成分之一。粘多糖的正确命名应该是糖胺多糖(glycosaminoglycan,GAG),它是由糖醛酸与乙酰氨基糖或其硫酸酯组成的二糖单位形成的重复序列。已知哺乳动物的GAG包括4-硫酸软骨素(chondroitin-4-sulfate,C4S),6-硫酸软骨素(C6S),硫酸皮肤素(dermatan sulfate,SD),硫酸角质(素)(Karatansulfate,KS)和肝素(haparin,HP)还有硫酸肝素或硫酸乙酰肝素(haparan,HS)总称为类肝素,及透明质酸(hyaluronic acid,HA)七种。这些糖胺多糖与核心蛋白连接后组成蛋白多糖(proteoglycan,PG)。蛋白多糖的旧名称粘蛋白。糖胺多糖就是酸性粘多糖(acid mucopolysaccharide,AMPS)。这些多糖的降解必须在溶酶体中进行,目前已知有10种溶酶体糖苷酶、硫酸酯酶和乙酰转移酶参与其降解过程,任何一种酶的缺陷都会造成氨基葡聚糖链的分解障碍而积聚体内,并自尿中排出。
病理改变
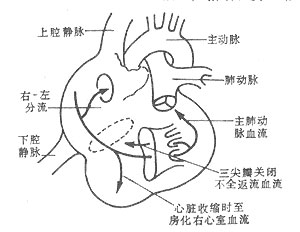
粘多糖在纤维细胞内沉积,染色成为气球样细胞,称为Hurler细胞,存在于肝、脾、淋巴组织的网状细胞中,在软骨细胞和成骨细胞,中枢神经系统和周围神经节,视网膜细胞和角膜细胞中也均有类似的物质堆积。在心内膜沉积形成斑状增厚,主动脉,肺动脉、冠状动脉和脑、肾、肝、脾和四肢的动脉壁均有沉积。
临床表现
出生后发育正常,1岁前逐渐出现体征。1岁后发育迟缓,骨骼畸形渐明显。头大,前额突出,颅骨呈舟状畸形。颈短,下胸部和上腰部脊柱后突。鼻梁扁平宽,嘴唇大而外翻,舌大张口,牙齿稀疏而小,牙龈肥厚。面容粗糙,表情淡漠,智能落后。毛发多粗糙而黑。多数有关节挛缩,掌、指宽而短,膝、髋外翻并有扁平足。腹部膨隆,肝脾肿大或有疝气,肝功能正常。角膜云翳混浊。鼻咽管畸形、耳聋,易发生中耳炎及上呼吸道感染和肺炎。胸廓畸形。由于粘多糖在血管壁的沉积,可突发冠状动脉闭塞或心肌梗塞。
实验室及其他检查
一、实验室检查
1、尿液粘多糖检测甲苯胺蓝呈色法常作为本病的筛查。阳性者用醋酸纤维薄膜电泳区分尿中排出的粘多糖类型,以协助分型。方法:收集晨尿,用吸液管将尿液0.1ml,一滴一滴地滴于滤纸上,使成6cm左右圆斑;(每滴一次尿后即用吹风机吹干)将已吹干的尿斑滤纸浸于0.2%甲苯胺蓝染液(甲苯胺蓝1g加蒸馏水100ml,再取该液5ml加丙酮20ml即成)染色45秒钟,取出使干,将上述已干的染色尿斑滤纸浸于10%醋酸中(冰醋酸10m加蒸馏水90ml)浸泡4分钟脱色,若不洁可再脱一次,空气中干燥。同时用正常人尿做对照。尿斑处呈紫蓝色环状或点状为阳性,正常人尿斑无色为阴性。
2、酶学分析各型MPS的确诊应依据酶活性测定,可采用外周血白细胞、血清或培养纤维细胞进行。
二、X线检查颅骨可见鞋形蝶鞍、颅骨呈舟状,颅板致密,颅缝早闭而前囟增大闭合延迟。鼻窦发育不良,充气少。下颌骨短且宽,髁突小而扁,关节凹浅。肋骨脊柱端细小,胸骨端增宽,形如“飘带”状。脊柱胸腰椎交界处后突成角状畸形。椎体畸形其前缘上方缺损,下方骨质如鸟嘴状突出,有的椎体发育不良,小而不规则,向后移位,尤以第1、2腰椎畸形最重。
长骨骨干的改变上肢较下肢明显,骨干粗而短,两端逐渐变细,骨皮质变薄。肱骨骨干“近端向背侧成角,尺挠骨”远端呈“V”形倾斜。股骨颈细长呈髋外翻,股骨头扁小致密。掌指骨短粗,远端宽,近端尖呈三角形,远节指骨呈爪形。指间关节挛缩,腕骨和跗骨骨化延迟且外形不规则。
诊断和鉴别诊断
一、粘多糖病I型
1、病因粘多糖病I型有2个亚型,均为α-1艾杜糖醛酸苷酶(α-Iduronidase)缺乏症,系因该酶的某种等位基因的突变所致。
粘多糖病I-H型(MPS-IH型),又称Hurler综合征,Hurler基因位于1号染色体上。在粘多糖中硫酸皮肤素和硫酸肝素中有L-艾杜糖醛酸的成分,其降解需要α-L-艾杜糖醛酸苷酶。由于此酶缺乏,其前体物的降解受阻而在体内堆积。硫酸皮肤素和硫酸肝素为角膜、软骨、骨骼、皮肤、筋膜、心瓣膜和血管结缔组织的结构成分,多为细胞膜外层的结构成分,细胞死亡后可释放出堆积的粘多糖。
2、诊断根据临床表现和X线骨片的改变,结合以下实验室检查可以诊断。
①末梢血白细胞,淋巴细胞和骨髓血细胞中可见到异染的大小不等、形状不同的深染颗粒,有时呈空泡状,颗粒称Reilly氏颗粒,经证实为粘多糖。
②患者尿中排出大量酸性粘多糖,③可超过100mg/24小时(正常为3~25mg/24h),确诊指标为证实尿中排出的为硫酸皮肤素和类肝素。患者白细胞,成纤维细胞或肝细胞和尿中缺乏α-艾杜糖醛酸酶。
3、鉴别诊断诊断时需与骨骼发育落后所致的矮小症相鉴别,如呆小症(先天性甲状腺功能减低症),多发性硫酸酶缺乏症(尿中硫化物和硫化胆固醇增多)。
二、粘多糖病Ⅱ型
1、病因粘多糖病Ⅱ型(unter syndrome)为X连锁隐性遗传。病因是艾杜糖醛酸-2-硫酸酯酶缺乏。临床上有重型(A)和轻型(B)。由于酶缺乏使硫酸皮肤素(DS)和硫酸类肝素降解障碍,在体内储留并由尿中排出,二者的排出量比为1:1.
2、临床表现临床上重型表现与粘多糖I-H型相似,多在青春期前死亡。起病在2~6岁,有特殊面容和骨骼畸形,但脊椎无鸟嘴样畸形。角膜内皮细胞虽有粘多糖沉积而无角膜云翳,皮肤呈结节性增厚以上臂和胸部为著。幼儿期始有听力损伤,呈进行性耳聋,视网膜变性,心脏增大可闻收缩期与舒张期杂音。最后可发生充血性心力衰竭或心肌梗塞,常是死亡的原因。智能落后的差异较大或严重或轻度落后。肝脏肿大,和关节强直。轻型无智能障碍,临床症状亦较轻。
3、诊断诊断依据尿中排出硫酸皮肤素与硫酸类肝素之比为1∶1.成纤维细胞培养,35S粘多糖积蓄,加入纯化的Hunter综合征因子可得到纠正,此可间接证明为艾杜糖硫酸酯酶活性缺乏。如能直接测血清和细胞内酶活性更可确诊。此类型可在产前测羊水细胞的酶活性,以指导计划生育。
三、粘多糖病Ⅲ型
1、病因粘多糖病Ⅲ型(Sanffilippo综合征)其特点为Ⅲ型有不均一性。其酶的缺乏各亚型不同。ⅢA型为硫酸酰胺酶(旧名称类肝素-N-硫酸酯酶)缺乏,ⅢB为α-N-乙酰葡糖胺酶缺乏,ⅢC为N-乙酰基转移酶缺乏,ⅢD为葡糖胺-6-硫酸酯酶缺乏,都是硫酸肝素降解所需要的酶,因此以上酶的缺乏均可引起硫酸(类)肝素(HS)在体内的蓄积,由尿中排出HS增多。此类酶缺乏主要引起神经系统不同程度的破坏,神经原呈汽球样变,脑室扩大,脑组织内硫酸类肝素、糖酯和GM-神经节苷脂含量增加,基底神经节损伤等。
2、临床表现在出生后一岁内精神运动发育正常。2~3岁时逐渐出现行为、语言等障碍,智能障碍,面容粗糙,关节强直和毛发过多。肝脾肿大。神经系症状表现为进行性手指徐动,四肢痉挛性瘫痪等。四种亚型的临床表现无区别,只ⅢA型临床进展较快。本型无角膜混浊,无心脏异常。
3、诊断根据尿中排出硫酸类肝素增多,甲苯胺兰试验常为阴性。分析成纤维细胞、白细胞和血清酶活性,可以确诊。临床上用P-硝基苯底物测定白细胞或血清的α-N-乙酰氨基葡糖苷酶,方法简单可靠。
四、粘多糖病Ⅳ型
1、病因粘多糖病Ⅳ型(Morquio氏病),有两个亚型。其病因为ⅣA为半乳糖-6-硫酸酯酶缺乏,ⅣB为β-D半乳糖酶缺乏。硫酸软骨素(CS)和硫酸角质素(KS)的降解障碍,而在细胞内沉积,硫酸角质素与软骨素-4/6-硫酸由尿中排出增多,但粘多糖总量不增多。随着年龄的增长硫酸角质素的浓度下降,至成年时排出量可正常。由于粘多糖在骨和软骨细胞沉积,骨发育障碍最为明显。Ⅳ型为常染色体隐性遗传。
2、临床表现Ⅳ型的临床特点为明显的生长迟缓,步态异常和骨骼畸形且逐渐显著。骨骼的畸形表现和I-S型相似,脊椎的鸟嘴突,椎骨扁平,飘带肋骨,还可有鸡胸,骨质疏松,髂骨外翻,股骨头变平,腕和膝关节肿大,但无关节强直。颜面呈颌骨突出,鼻矮,口大、牙间隙宽及牙釉质发育不良。学龄期出现角膜混浊,皮肤增厚且松弛。智力发育基本正常为Ⅳ型的特点。青春期发育可正常。逐渐出现脊髓压迫症状,晚期出现麻痹性截瘫和呼吸麻痹。病人寿命多为20~30岁。
3、诊断需测尿中粘多糖和测白细胞等组织细胞酶活性。
五、粘多糖病Ⅵ型
1、病因粘多糖病Ⅵ型又称Maroteaux-Lamy综合征。为N-乙酰半乳糖胺-4-硫酸酯酶缺乏,临床上分重型和轻型。本型为常染色体隐性遗传,基因在5号染色体长臂5q13.3区。酸性粘多糖以硫酸皮肤素(DS)沉积为主,约占尿排出酸性粘多糖的70%~95%,其余可能为硫酸软骨素和硫酸类肝素。
2、临床表现临床重型表现多从2~3岁开始生长迟缓,关节活动严重受限,颈短,角膜混浊发生较早,颅骨蝶鞍呈鞋型,颅骨缝早闭合可引起神经系症状,出现脑积水和痉挛性偏瘫。骨骼畸形的程度个人间差异较大,逐渐发生骨骼畸形如I-H型上肢长骨受累比下肢重。可有肝脾肿大。智力正常,但可有眼失明和耳聋。心脏亦可有异常可引起死亡,寿命多不超过10岁。
3、诊断诊断依尿中排出酸性粘多糖以硫酸皮肤素为主,分析白细胞的酶活性可以确诊。
六、粘多糖病Ⅶ型
1、病因粘多糖病Ⅶ型是β-D-葡糖醛酸酶缺乏,为常染色体隐性遗传,该酶基因位于7q21.2-q22区。Ⅶ型临床上少见。
2、临床表现在出生后不久即出现特殊面容,眼距宽,鼻梁低平,上颌骨突出,眼内眦赘皮小。骨骼畸形可有鸡胸和鸟嘴形脊椎启弯,椎体扁平。上肢短,骨骼发育增速,皮肤粗糙,而松弛,肝脾肿大逐渐加重。神经系损伤不明显。主动脉可有缩窄。
3、诊断根据临床和尿中排出酸性粘多糖增多。确诊需测定组织细胞和血清,尿液中缺乏β-D-葡糖醛酸酶活性。羊水细胞培养后测酶活性可以产前诊断。
七、粘多糖病Ⅷ型
1、病因粘多糖病Ⅷ型1978年开始报道,病因是由于N-乙酰氨基葡糖-6-硫酸酯酶缺乏,体内蓄积大量的硫酸角质素(KS)和硫酸类肝素(HS),二者在尿中以3∶1的量排出。
2、临床表现有粘多糖病Ⅲ型和Ⅳ型的共同特征,有侏儒,智能落后,脏器受累和骨骼畸形,无角膜混浊。
3、诊断诊断依据尿中排出酸性粘多糖为KS和HS且呈3∶1的量排出,确诊需测细胞或血清酶活性。
八、粘多糖病边缘性疾病近年来由于生物化学以及酶的代谢方面的不断深入研究,又发现了一些异于上述六型的粘多糖病边缘性疾病,其症状与粘多糖病类似,但尿中排出粘多糖不增加,简述如下:
1、类风湿型粘多糖病:
Winchester等于1969年发现两例同胞病儿,他们的临床表现与粘多糖病I(H)型相似,而骨骼变化似类风湿性关节炎,X线照片表现为进行性溶骨性破坏,尿中排出的粘多糖量正常。通过皮肤纤维母细胞组织培养证实为粘脂质代谢障碍。
2、甘露糖累积病:
Kjellman等于1969年发现1例临床表现很似粘多糖病I(H)型而X线骨骼病变很轻微,生化检查发现病儿肝内缺乏α-甘露糖酶,造成甘露糖代谢障碍以致大量沉积于中枢神经系统。男性较多。均有骨骼变化和智力发育延迟。
3、岩藻糖或去氧半乳糖累积病(fucosidosis):
Durand等于1966年报告两例同胞兄妹(年龄为3岁和4岁)表现为进行性智力发育障碍,脊柱变形,肌力减低,进行性痉挛和去大脑皮质性强直,消瘦,皮肤变厚,大量出汗,心脏增大以及经常发生呼吸道及中耳感染。其生化的基本变化是缺少α-L-去氧半乳糖酶,造成皮肤、淋巴细胞以及其他组织积累糖脂,是一种神经内脏的累积病。为常染色体隐性遗传。
4、粘脂质累积病I型(mucolipidosisI):
症状和骨病变很象Hurler综合征,但较轻,且进展缓慢,以后出现肌张力减低,共济失调及周围神经症状,年长儿可有惊厥。无角膜混浊。周围血淋巴细胞和骨髓细胞有空泡形成或颗粒。肝内β-半乳糖苷酶的活性增高。遗传方式为常染色体隐性遗传。
5、粘脂质累积病Ⅱ型(mucolipidosisⅡ):
又称包涵体细胞病,Leroy氏于1969年报告两例,他们的临床表现和X线所见与粘多糖病I(H)型相似外,还好发髋关节脱位,而尿中粘多糖的排出量是正常的。皮肤组织培养发现纤维母细胞胞浆内有黑色的包涵体,因此称为包涵体细胞病。为常染色体隐性遗传。
6、粘脂质累积病Ⅲ型(mucolipidosisⅢ):
又称Pseudo-Hurler polydystrophy,临床表现和骨骼变化与粘多糖病I(H)型或Ⅱ型相似,有些病人可见髋关节脱位,头颅表现正常。内脏和间质组织中有糖脂和粘多糖累积,尿中粘多糖的排出量正常。为常染色体隐性遗传。
治疗
该病一般无特殊治疗。特异的治疗方法是骨髓移植,以替代粘多糖病各型酶的缺乏。酶替代和基因治疗正在研究中。各型粘多糖病大部分可以进行羊水细胞cDNA基因分析作产前诊断。